Hola a continuación les presento mi informe acerca de la nitración del benzoato de metilo. Espero les sirva de guía o aclare algunas dudas que tengan. Las imágenes utilizadas fueron sacadas de las referencias bibliográficas mencionadas o creadas con el programa ChemSketch.
<center>Universidad Simón Bolívar
Departamento de Química
Sección de Química Orgánica
Laboratorio de Química Orgánica para Ingenieros
PRÁCTICA V: NITRACIÓN DEL BENZOATO DE METILO
Kevin Mejías 11-10600
</center>
RESUMEN
Se realizó la nitración del benzoato de metilo a través de una sustitución electrofílica aromática. Se hizo reaccionar primero ácido sulfúrico concentrado con benzoato de metilo y luego se agregó nitrato de potasio. Se obtuvo como producto meta-nitrobenzoato de metilo. La cromatografía de capa fina del benzoato de metilo y el m-nitro benzoato de metilo aplicando una solución de hexano-acetato de etilo (9:1) se obtuvieron los siguientes valores de relaciones de frente (Rf): 0,79 y 0,42 respectivamente.
INTRODUCCIÓN
La reacción más importante de los compuestos aromáticos es la sustitución electrofílica aromática. En este tipo de reacción, un hidrógeno del anillo aromático es remplazado por un electrófilo (E+), de ahí el nombre de sustitución aromática electrofílica. Son varios los tipos de sustituyentes que se pueden añadir al anillo aromático, sin embargo, en la práctica se realizara la nitración del benzoato de metilo. En esta reacción se sustituye un hidrógeno por un grupo nitro , que se ubica en posición meta al grupo del éster del benzoato [1].
Para generar el electrófilo necesario, el ácido sulfúrico protona al ácido nítrico, que pierde
agua y forma un ion nitronio, el electrófilo necesario para la nitración [1].
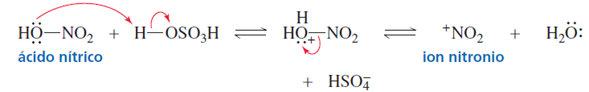
Figura 1. Obtención del ion nitronio a partir de la protonación del ácido nítrico.
Después se lleva a cabo la reacción de nitración
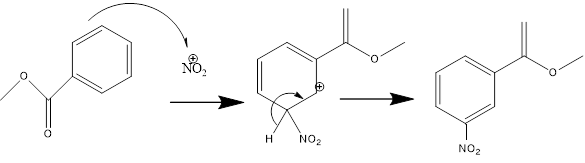
Figura 2. Mecanismo de reacción de la nitración del benzoato de metilo.
En la reacción, es posible obtener como impurezas en la reacción pequeñas cantidades de
los isómero “orto” y “para” del m-nitrobenzoato de metilo. Estos productos secundarios se eliminan cuando el producto deseado se lava con metanol y se purifica por cristalización. El agua tiene un efecto retardante sobre la nitración porque interfiere con los equilibrios ácido nítrico-ácido sulfúrico que forman los iones de nitronio. Cuanto menor es la cantidad de agua presente, más activa es la mezcla de nitración. Además, la reactividad de la mezcla de nitración puede controlarse variando la cantidad de ácido sulfúrico utilizado. Este ácido
debe protonar al ácido nítrico, que es una base débil. Cuanto mayor sea la cantidad de ácido disponible, más numerosas serán las especies protonadas. El agua interfiere porque es una base más fuerte que H2SO4 y HNO3. La temperatura también es un factor para determinar el grado de nitración. Cuanto mayor es la temperatura, mayores serán las cantidades de productos de dinitración formadas en la reacción [2].
La técnica de cromatografía se basa en el principio general de distribución de un compuesto entre dos fases, una fija y otra móvil. Así, la cromatografía se puede considerar como una remoción selectiva de los componentes presentes en una mezcla por acción de la fase móvil que fluye a través de la fase fija, donde se encuentran. Esta técnica no sólo es útil en separaciones, también sirve como indicador del grado de pureza e identificador de una sustancia [3].
Existen varios tipos de cromatografía, entre ellas están la cromatografía por adsorción, de partición, cromatografía de filtración con geles y cromatografía de intercambio iónico. Adicionalmente existen también varias técnicas asociadas a los tipos de cromatografía mencionados anteriormente, entre ellas están, la cromatografía de capa delgada, cromatografía en capa preparativa, cromatografía de columna, cromatografía de papel y la cromatografía gaseosa. No importa qué tipo de cromatografía se emplee, todas conllevan los mismos 5 pasos iniciales. El primer paso es el armado de la placa o columna, el cual implica la disposición espacial que adoptará la fase estacionaria. El segundo paso es la siembra de la muestra, se refiere al contacto inicial de la mezcla a separar o analizar con la fase estacionaria, para su posterior desarrollo. El tercer paso es el desarrollo, se trata de la transición de la fase móvil a través de la fase estacionaria. El cuarto paso se conoce con el nombre de revelado, que consiste en la localización de las zonas en las que se encuentras los compuestos ya separados. Por último, el quinto paso se le conoce como elución, se aplica cuando se quiere remover los solutos de la fase estacionaria [3].
En la práctica se empleará una cromatografía de adsorción haciendo uso de la técnica de capa delgada. La cromatografía de adsorción se basa en la separación de un soluto entre una fase sólida de adsorbente y una fase móvil. El fenómeno de adsorción ocurre en la superficie del granulo en la fase fija, y se fundamenta en la tracción entre el soluto y el adsorbente por formación de uniones dipolo-dipolo y formación de puentes de hidrógeno. El adsorbente es un sólido que tiene la capacidad de contener en su superficie un componente presente en una fase liquida o gaseosa. Así la cromatografía de adsorción es una adsorción solido líquido y depende de dos factores. El primero es el equilibrio establecido en la interface entre el sólido adsorbido y la solución aplicada como fase móvil. El segundo factor es la solubilidad relativa del soluto en la fase móvil. De manera que el soluto es adsorbido en la superficie del adsorbente y luego desorbido por el solvente en la fase móvil [3].
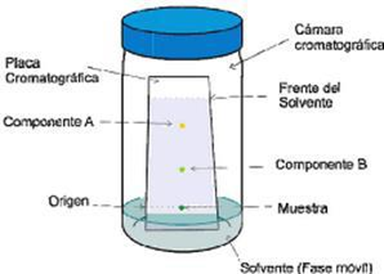
La cromatografía de capa delgada es de aplicación analítica se utiliza comúnmente para seguir el desarrollo de una reacción, se observa la formación de productos y la separación de reactivos, para así poder analizar el número aproximado de componentes en una muestra. También tiene gran valor como criterio de pureza e identificación de sustancias. Otra utilidad que tiene la técnica de capa delgada es la de determinación de solvente de desarrollo adecuado para una cromatografía de paca preparativa o para elegir la secuencia de solventes apropiados para la elución de los componentes en una mezcla. A continuación, se presenta un esquema del proceso de la cromatografía de capa delgada [4].
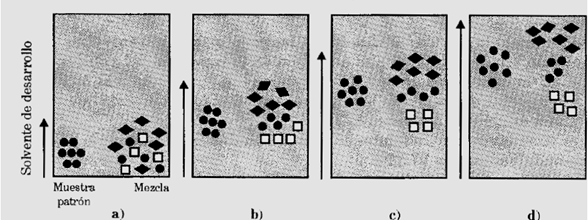
Figura 3. Esquema de la cromatografía de capa delgada. Figura obtenida de [3].
En la figura anterior se muestra como se ha sembrado una muestra y el patrón de un posible componente en la parte inferior de la capa adsorbente, Luego se sumerge dicha capa en un solvente de desarrollo, que asciende por capilaridad, desorbiendo selectivamente a los distintos componentes de la mezcla pasando por los estados “a”,” b”,” c” y “d”. Ver figura 3 [3].
Del gráfico se puede concluir que  fue la sustancia mas retenida y la más móvil fue la sustancia  . La sustancia  se desplazó de forma intermedia similar a la sustancia patrón, por lo tanto, se podría tratarse de la misma sustancia [3].
Se puede realizar un estudio análogo al mencionado previamente de forma microscópica.
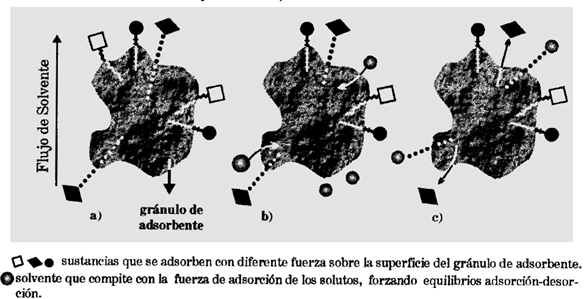
Figura 4. Analogía microscópica de la cromatografía TLC. Figura obtenida de [3].
En la figura 4 se puede observar un granulo adsorbente el cual ha adsorbido 3 compuestos, cada uno con diferente fuerza, de manera que,  . Al comenzar la elución. Ver figura 4.b, las moléculas de solvente competirán con los solutos por su adsorción en la superficie del gránulo adsorbente. El compuesto  es el adsorbido más débilmente y por tanto es el más fácil de desorber. Entonces el solvente de desarrollo lo remueve, arrastrándolo a medida que avanza sobre la fase estacionaria. Ver figura 4.c.
De forma que las fuerzas con las que los solutos se fijan al adsorbente resultan de la polaridad de los mismos. Un solvente de desarrollo muy polar, tiene una fuerza de elución grande y remueve a su paso la muestra que ya no queda ligada al adsorbente. Entonces para un desarrollo en el que dos o más componentes corren con el frente de solvente, no sirven para fines separativos.
El desplazamiento alcanzado por cada componente de la mezcla y por el patrón sembrados se puede medir a través de la relación de frente “Rf”, este se define matemáticamente por:
Rf=b/a
Rf=Distancia recorrida por el compuesto de estudio)/(Distancia recorrida por el solvente de desarrollo)
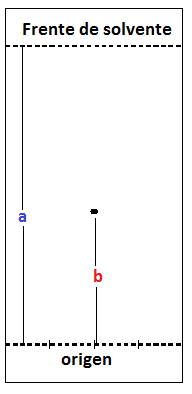
Figura 5. Figura ilustrativa de las variables presentes en el cálculo de Rf en una cromatografía TLC.
Entonces hablar de Rf es hablar de la movilidad de un compuesto o en un proceso cromatógrafico. Las variables generales que afectan la movilidad en una experiencia de cromatografía por adsorción son el adsorbente, estructura del soluto, solvente de desarrollo, temperatura y la saturación de la cuba. Algunos de estos factores serán discutidos a continuación. La estructura del soluto está relacionada con su habilidad para ser adsorbido y desorbido y depende de si polarizabilidad y de la capacidad de formar puentes de hidrógeno. Por esta razón los grupos polares son adsorbidos más fuertemente. En general el poder de ser adsorbido aumenta con el incremento de grupos funcionales unidos a la misma molécula. Sin embargo, existen casos en los cuales dos grupos funcionales presentes pueden formar uniones de hidrógenos intermoleculares y en consecuencia adsorben menos. El solvente de desarrollo es de vital importancia para tener éxito en un proceso separativo. Si el solvente es muy polar, será fuertemente adsorbido por los gránulos de fase fija y desplazamiento por adsorción será inmediato. El resultado de esta desorción indiferenciada será que la mayoría de los solutos correrán con el frente de solvente y no se logrará separación alguna. Debido a esto es que es una condición general empezar el desarrollo con un solvente de baja polaridad para luego aumentarla gradualmente. Otra variable es la velocidad de elución, la cual debe ser lenta para permitir que se tienda al equilibrio adsorción-desorción. La elución se ve influenciada por la temperatura, la cual a su vez afecta a la volatilidad y a la solubilidad de una sustancia. Por lo tanto, la temperatura debe mantenerse constante en una cromatografía por adsorción [3].
En la práctica se realizó la cromatografía de capa delgada para determinar el grado de pureza del m-nitrobenzoato de metilo sintetizado. Adicionalmente se realizó también la cromatografía de capa fina a las aguas madres de la reacción antes mencionada y a una muestra de benzoato de metilo con la finalidad de comparar dichas cromatografías y analizar los resultados obtenidos.
OBJETIVOS
*Realizar la nitración del benzoato de metilo por medio de una reacción de sustitución electrofílica aromática.
*Determinar el grado de pureza del cristalizado mediante la técnica de cromatografía de capa delgada.
SECCIÓN EXPERIMENTAL
A continuación, se presenta un esquema gráfico de la nitración del benzoato de metilo
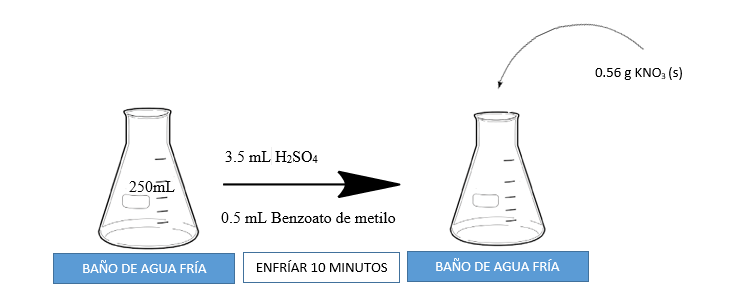
La mezcla se agita para ayudar a disolver el KNO3 (s) y se deja enfriar por otros 10 minutos. Luego se retira la mezcla del baño de agua fría y se deja atemperar. Una vez la mezcla se encuentre a temperatura ambiente se filtra al vacío. Ver la siguiente figura.
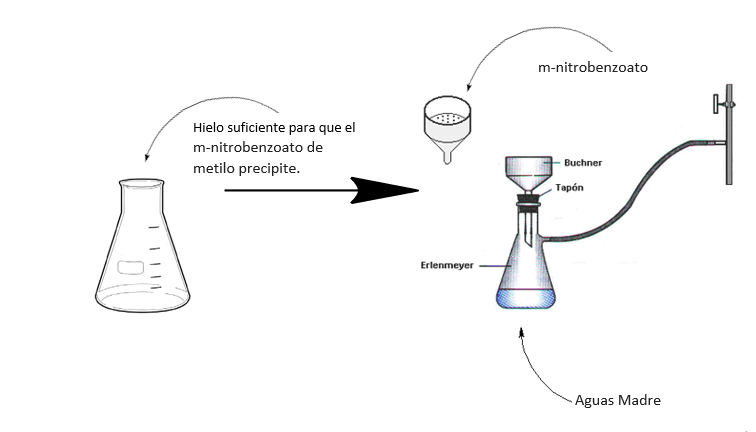
Luego se procede a sembrar las muestras de m-nitrobenzoato de metilo sintetizado, aguas madres y benzoato de metilo con unos capilares muy finos en las placas de TLC.
Una vez finalizada la cromatografía, se expuso la placa de TLC a una luz ultravioleta y se marcaron con un lápiz de grafito las marcas que se pudieron observar. El solvente utilizado fue una Solución de hexano-acetato de etilo 9:1.
RESULTADOS Y DISCUSIÓN
Tabla 1. Rf obtenidos de la cromatografía TLC realizada
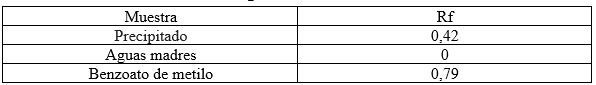
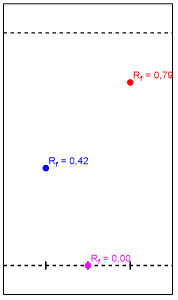
Figura 6. Esquema gráfico de los Rf obtenidos.
Las manchas correspondientes al benzoato de metilo y el precipitado se pueden diferenciar por su polaridad. El solvente empleado, hexano-acetato de etilo 9:1, es apolar y la placa un sólido adsorbente polar. De manera que el precipitado al tener una mayor afinidad a la fase estacionaria que el benzoato de metilo se retuvo más en la placa. Esto ocurre porque el grupo nitro presente en la sustitución electrofílica, hace el precipitado adquiera cierta polaridad relativa sobre el anillo bencénico, de modo que es más polar que el benzoato de metilo.
Para poder apreciar los resultados de la técnica de la cromatografía de capa fina, se colocó la placa en rayos UV debido a que la longitud de onda que transmiten los compuestos no son perceptibles al ojo humano, pero que al activarlos con la luz UV si se hacen visibles.
CONCLUSIONES
Se presume que se sintetizó con éxito m-nitrobenzoato de metilo debido al color y las características físicas del precipitado que se pudieron apreciar. Entre ellas un olor característico a menta del m-nitrobenzoato de metilo. La cromatografía TLC concuerda con la teoría de capa de adsorción y con la teoría de los factores que afectan la movilidad. De forma que Rf Benzoato de metilo> Rf m-nitrobenzoato de metilo > Rf Aguas madres.
BIBLIOGRAFÍA
1. Paula Yurkanis Bruice. “Química Orgánica”. Pearson Prentice Hall. (2008).
2. Donal L. Pavia, Gary M. Lampman, George S. Kriz y Randall G. Engel. “A small Scale Approach to Organic Laboratory Techniques”. Brooks/Cole (2011).
3. Lydia Galagovsky Kurman. “Química Orgánica. Fundamentos teórico-prácticos para el laboratorio”. Eudeba. Primera Edición.
4. Joseph Sherma y Bernard Fried. “Thin layer chromatography”. Marcel Dekker,Inc.1996